19-06-04 Pape, Katrin, 2019¶
Pape, K., Tamouza, R., Leboyer, M. and Zipp, F., 2019. Immunoneuropsychiatry—novel perspectives on brain disorders. Nature Reviews Neurology, p.1.
Contents¶
- 00. Abstract
- 01.
- 02. Neuroimmune interplay in brain health
- 03. Cognitive performance in inflammation
- 04. Autoimmunity
- 05. Immunopsychiatry - an emerging field
- 06. Immunomodulatory treatment
- 07. Resilience and reserve
- 08. New clinical approach
- 09. Conclusion
- 10. References
00. Abstract¶
- classical neuroinflammatory disease
- MS
- autoimmune encephalitis
- psychiatric diseases such as
- schizophrenia
- autism spectrum disorder
- bipolar disorder
- depression
pathways:
- microglial activation
- pro-inflammatory cytokines
- molecular mimicry
- anti-neuronal autoantibodies
- self-reactive T cells
- disturbance of the blood–brain barrier
Immune processes have a vital role in CNS homeostasis, resilience and brain reserve. Our cognitive and social abilities rely on a highly sensitive and fine-tuned equilibrium of immune responses that involve both innate and adaptive immunity. Autoimmunity , chronic inflammation, infection and psychosocial stress can tip the scales towards disruption of higher-order networks. However, not only classical neuroinflammatory diseases, such as multiple sclerosis and autoimmune encephalitis, are caused by immune dysregulation that affects CNS function. Recent insight indicates that similar processes are involved in psychiatric diseases such as schizophrenia, autism spectrum disorder, bipolar disorder and depression. Pathways that are common to these disorders include microglial activation, pro-inflammatory cytokines, molecular mimicry , anti-neuronal autoantibodies, self-reactive T cells and disturbance of the blood–brain barrier. These discoveries challenge our traditional classification of neurological and psychiatric diseases. New clinical paths are required to identify subgroups of neuropsychiatric disorders that are phenotypically distinct but pathogenically related and to pave the way for mechanism-based immune treatments. Combined expertise from neurologists and psychiatrists will foster translation of these paths into clinical practice. The aim of this Review is to highlight outstanding findings that have transformed our understanding of neuropsychiatric diseases and to suggest new diagnostic and therapeutic criteria for the emerging field of immunoneuropsychiatry.
01.¶
Crosstalk between the immune and nervous systems is receiving increasing attention in a wide spectrum of neurological and psychiatric diseases. As pathways emerge that are common to disorders from both fields, the traditional boundaries between neurological and psychiatric disorders are becoming blurred. Novel discoveries about the roles of the immune system in CNS function and in disease together with tremendous developments in immune therapies make this topic of great interest. This rapidly developing research is providing new perspectives not only on disease and therapeutic targets but also on brain reserve and resilience in neuropsychiatric disorders.
In this Review, we first give an overview of neuro-immune interplay and inflammatory influences in the healthy brain and in disease. Accumulating data suggest immune and autoimmune contributions to a wide variety of neurological and psychiatric disorders (BOX 1). These findings are reflected in a growing number of therapeutic studies of mechanism-based immune treatments in subgroups of patients with neurological and psychiatric disorders. In addition, we discuss recent insights into the role of psychosocial stress and infectious events in the CNS that provide a mechanistic cornerstone for our understanding of neuroinflammatory processes and brain diseases. On the basis of the find-ings and studies discussed, we suggest a new clinical approach to neuropsychiatric disorders from an immunological perspective.
Box 01. Neuropsychiatric disorders with inflammatory disturbance¶
Involvement of immune dysfunction in pathogenesis has been studied in a broad range of neuropsychiatric diseases. This Review focuses on the following disorders:
- Multiple sclerosis
- Neuromyelitis optica spectrum disorder
- Autoimmune encephalitis (paraneoplastic, idiopathic or triggered by infection)
- Schizophrenia
- Autism spectrum disorders
- Depression
- Bipolar disorder
- Dementia, especially Alzheimer disease
In addition, immune mechanisms are likely to contribute to many other disorders. These disorders include the following:
- Myelin oligodendrocyte glycoprotein antibody spectrum disorder
- Chronic inflammatory optic neuritis
- Rasmussen encephalitis
- Susac syndrome
- Stroke
- Parkinson disease
- Amyotrophic lateral sclerosis
- Huntington disease
- Obsessive–compulsive disorder
- Anxiety disorders
- Eating disorders
02. Neuroimmune interplay in brain health¶
interplay between neurons, glial cells, immune system conntributes to functional propertyies
The CNS has traditionally been regarded as a site of immune privilege. However, despite being protected by specialized physical barriers, the brain is neither inert nor immunologically separated from the peripheral immune system. Instead, an interplay between neurons, glial cells and the immune system contributes to functional properties, such as cognition, social behaviour and learning performance in the healthy brain.
- micloglia
- chemokine / chemokine receptor
- extracellular vesicles / signaling modelecules
Increasing evidence indicates that specific commu-nication occurs between neurons and microglia, the brain parenchyma-resident macrophages that account for ~10% of CNS cells. Microglia permanently survey their local environment with fine, motile processes[1] and, during brain development, they have an impor-tant role in synaptic pruning mediated by the comple-ment proteins C1q and C3, as they phagocytose the complement-tagged synapses[2],[3]. In addition to micro-glia, soluble factors such as chemokines and chemokine receptors also contribute to physiological developmental processes in the CNS; for example, CXC-chemokine receptor 4 (CXCR4) and CXCR7 are involved in the migration of cortical interneurons4. In the healthy adult brain, microglia have a role in the homeostasis of synaptic circuits in the CNS[5]. Their expression of receptors for purines (such as ATP) and common neurotransmitters (such as glutamate) enables them to sense local neuronal activity[6]. In response, microglia can directly contact neurons via outgrowth of processes5 or can indirectly modulate neuronal firing rate via release of extracellular vesicles[7] or signalling molecules, such as tumour necro-sis factor (TNF)[8]. In this way, microglia contribute to activity-induced synaptic plasticity, for example, during motor learning and memory. During systemic inflam-mation, microglia can become activated and produce pro-inflammatory mediators and induce phagocytosis9.
Microglia comprise the innate CNS immune compartment; the existence of adaptive immune cells in the brain has long been considered a sign of disease. However, in healthy individuals, antigen-presenting cells and T cells patrol the brain’s borders, residing in the meninges, the choroid plexus and the cerebro spinal fluid (CSF)10. These patrol cells provide additional protection to that provided by the blood–brain barrier (BBB) — a physical boundary that consists of the basal lamina of endothelial cells, tight junctions between them and astrocyte end-feet processes — and the blood–CSF barrier at the choroid plexus[11], which is composed of epithelial cells[12]. Notably, evidence suggests that the choroid plexus has a role not only in transmigration but also in stimulation of T cells in response to peripheral inflammatory signals13.
- maintainance of neuronal fn
Although T cells do not generally penetrate the parenchyma in non-inflammatory conditions, they can release soluble cytokines that affect CNS function[14],[15]. Studies in mice have demonstrated that adaptive immunity is necessary for cognitive performance: mice with severe combined immune deficiency exhibited impaired spatial learning and memory, but symptoms were reversed by injection of exogenous T cells16. Similar observations have been made for social behaviour14. From a neuro-biological perspective, levels of neurogenesis in adult transgenic mice that overexpress a CNS-specific T cell receptor are higher than those in mice that overexpress a T cell receptor for a non-CNS-specific antigen[17]. The advantageous effects of self-reactive T cells on the maintenance of neuronal function have led to the concept of protective autoimmunity, in which adaptive immune cell function maintains tissue function. Further beneficial roles of the immune response in the CNS — again going against the anticipated pathogenic activity of inflamma-tion — include protective roles for T helper 2 (TH2) cells in CNS injury and for IL-4 signalling in neurons in mul-tiple sclerosis (MS) models[18],19. The complex and diverse roles of adaptive immune cells are further underlined by the finding that a lack of B cells does not impair learning behaviour in mice[20].
drainage system
- glymphatic system (glial-dependent perivascular network)
- meningeal lymphatic system
The interplay between the CNS and the periphery is mediated by two drainage systems. The first is the glymphatic system, a glial-dependent perivascular network that ensures provision of nutrients for neurons and glia and clearance of extracellular metabolites, such as lactate[21]. Activation of the glymphatic system is higher during sleep than during wakefulness, underlining the impor-tance of sleep for removal of potentially neurotoxic waste products22. The second is the meningeal lymphatic system that lines the dural sinuses and allows drainage from the CNS to deep cervical lymph nodes, the discovery of which established the missing link to the peripheral immune system23,[24]. Via this system, molecules and immune cells from the CNS can be transported to lymphoid organs and evoke an immune response that involves subsequent migration of immune cells to the brain[25].
03. Cognitive performance in inflammation¶
03.00.¶
As described above, innate and adaptive immunity are essential for CNS homeostasis. Consequently, distur-bances in the equilibrium of immune cells, neurons and glial cells needed for healthy CNS function are likely to modify cognitive performance (FIG. 1a). In this section, we consider the evidence that low-grade inflammation and inflammation in response to infection can alter cognitive performance.
Recognition of pathogen-associated or damage- associated molecular patterns and subsequent phago-cytosis or cytokine production by microglia or invad-ing macrophages, representing the innate immune response in the CNS, has to be distinguished from the antigen-specific responsiveness of adaptive immune cells.
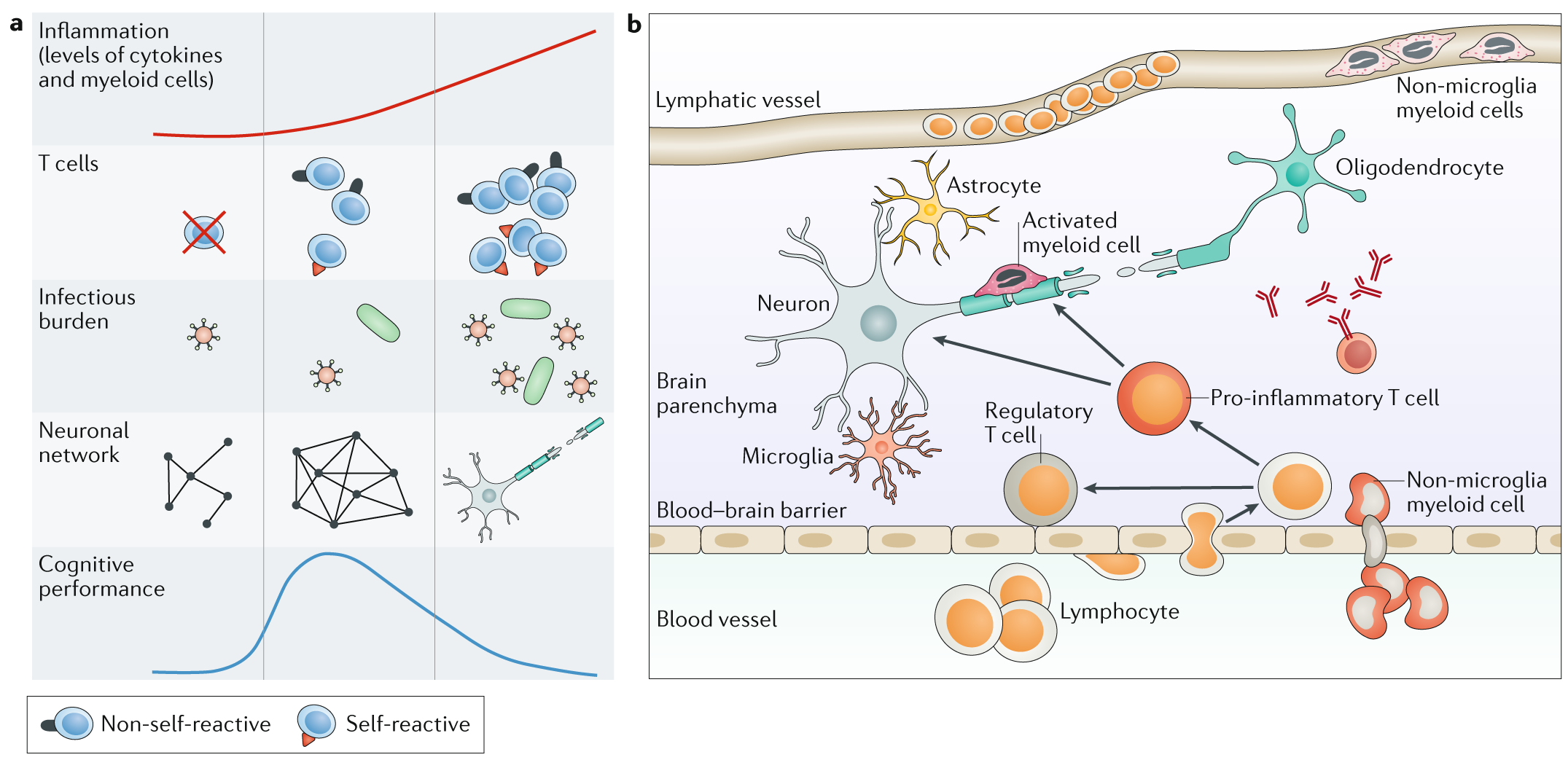
Fig. 1 | The interplay between the immune system and the CNS.¶
a | A certain level of inflammation and autoimmunity is necessary for optimal function of the CNS, but an overwhelming immune reaction leads to neuronal loss and impaired cognition.
b | Immune cell infiltration into the CNS in disease. Glial cells, including microglia and astrocytes, are resident in the CNS. Lymphocytes and non-microglia myeloid cells are both thought to be present in the lymphatic vessels in health but might transmigrate through the blood–brain barrier in inflammatory diseases.
03.01. Low-grade inflammaiton¶
sickness behavior
euflammation
A compelling example of the pathogenic influence of the immune system on brain function is the change in mood, social behaviour and cognitive abilities — known as sickness behaviour — upon infection and systemic inflammation. The release of pro-inflammatory cytokines, such as IL-1β, IL-6 and TNF, outside the CNS affects the brain via neural (mainly vagal) pathways, interaction with cytokine receptors on cerebral endothelial cells and/or microglial activation26. The behavioural effects, such as social with-drawal and fatigue, are assumed to be adaptive responses that increase survival of the host27. Similarly, treatment with the cytokine IFNβ, for MS or chronic viral hepa-titis, for example, has been associated with depressive symptoms as an adverse effect[28]. Sickness behaviour can be diminished after repeated subthreshold exposure to pathogens, a mechanism that is referred to as euflammation and that evidence suggests is the result of tolerogenic processes[29].
inflammaging
In contrast to this transient low-grade inflammation, chronic low-grade inflammation can occur and cause neurotoxicity and neurodegeneration[30]. For example, cytokines can have neurotoxic effects by increasing pro-duction of reactive oxygen species, reducing monoamine transmission and potentiating glutamatergic transmis-sion. Chronic, sterile, low-grade inflammation occurs during human ageing and can contribute to age-related diseases; this process is referred to as inflammaging31. Chronic inflammation is implicated in schizophrenia and other psychiatric disorders32, and systemic inflammation, as well as acute infections, have been associated with an increased rate of cognitive decline and exacerbation of symptoms in patients with Alzheimer disease (AD)[33]. Chronic low-grade inflammation is also present in people who are obese[34], and this inflammation influences cognitive performance by damaging neuronal circuits and the BBB and by activating pro-inflammatory immune cells[35]. These obesity-related mechanisms were initially observed in the hypothalamus, but some evidence sug-gests that they can also occur in the hippocampus, cortex, brainstem and amygdala[36].
- beneficial
- euflammation
- sickness behavior
- adverse
- chronic inflammation ⇒
- neuronal damage
- cognitive decline
- dementia
- chronic inflammation ⇒
∴ right balance of inflammation
Overall, in the interplay between the brain and the immune system, inflammation is a double-edged sword. Beneficial effects such as euflammation and evolution-arily advantageous sickness behaviour are on one side, but chronic inflammation that leads to neuronal dam-age, cognitive decline and possibly dementia is on the other side. The right balance of inflammation is needed for optimal CNS function.
03.02. Infection-related inflammation¶
Sydenham chorea
paediatric autoimmune neuropsychiatric disorders associated with S. pyogenes (PANDAS)
Infection can initiate chronic inflammation that alters cognitive function. Data from a Danish longitudinal register showed that prior hospitalization for an autoimmune disease or infection increased the risk of a major mood disorder by 45% and 62%, respectively[37]. In rodents, even infection and systemic inflammation in the fetus during the prenatal or perinatal period can cause long-term cognitive damage, including learning, memory and attention abnormalities, a model that explains why early infection increases the risk of psychosis in young adulthood[38],[39]. Similarly, a viral infection in the mother during the first trimester of pregnancy and bacterial infection during the second trimester of pregnancy were associated with the development of autism spectrum disorder (ASD), a pervasive neurodevelopmental disorder defined by impairments in social skills and stereotypical behaviour, among the children in a large Danish cohort[40].
In children, throat infection with Streptococcus pyogenes has repeatedly been associated with subsequent neuropsychiatric disorders[41]. In particular, these disorders include Sydenham chorea, a movement disorder related to rheumatic fever that occurs in temporal relationship to group A streptococcal infection[42], and conditions encompassed by the umbrella term of paediatric autoimmune neuropsychiatric disorders associated with S. pyogenes (PANDAS) and concomitant with obsessive–compulsive disorder, tic disorder or choreiform motoric hyperactivity[43]. The suspected pathophysiology of PANDAS is cross reactivity of anti-streptococcus A antibodies with brain tissue due to molecular mimicry[44] that initiates an adaptive immune cell response. Nevertheless, in a prospective study of children with post-streptococcal neuropsy-chiatric symptoms, no correlation was seen between clinical symptoms and a change in autoimmune markers, such as anti-neuronal antibodies or inflam matory cytokines[45]. Therefore, despite confirmation on an epide miological level, the aetiological association between streptococcal infection and psychiatric symptoms is still controversial and the subject of an ongoing, large observational study[46].
Severe infections, such as herpes simplex virus (HSV) encephalitis at any time in the lifespan, have been associated with long-term impairment of memory and brain atrophy[47]. Herpes family viruses persist in the host and require constant immune surveillance in order to prevent reactivation. For example, latent cytomegalovirus (CMV) infection leads to accumulation of functionally exhausted effector T cells while the naive T cell pool is diminished[48]. In the prospective Northern Manhattan Study, a high infectious burden (assessed with serological markers for Chlamydia pneumoniae, Helicobacter pylori, CMV, HSV-1 and HSV-2) was associated with cognitive decline independently of cardiovascular risk among a cohort of 1,625 patients[49]. Evidence suggests that among patients with schizophrenia or bipolar disorder, infections with Toxoplasma gondii, HSV and CMV affect the cognitive dysfunction already present in these disorders, in particular working memory[50]. Another example of the intricate relationship between cognition and chronic infection is HIV-associated neurocognitive disorder, which emerges in patients with HIV infection despite highly active antiretroviral therapy[51].
Our understanding of the mechanistic links between infectious burden and neuropsychiatric diseases is still in its infancy. In addition to molecular mimicry that triggers autoimmune reactions, a possible mecha-nism is stress-induced potentiation of microglial inflammasome activation, which causes an increase in pro-inflammatory mediators[52],[53]. In this way, both innate (microglial activation with subsequent phagocytosis or cytokine production) and adaptive immune responses (antigen-specific responsiveness and production of anti-bodies) contribute to pathology. In addition, infectious agents might act via epigenetic pathways to modulate the innate immune cell repertoire, thereby influencing the risk profile for neurodegenerative disorders[54].
In addition to the cumulative effect of infectious burden, pathogens contribute directly to neurodegen-eration, and therefore cognitive decline, in elderly people. One example that has been investigated extensively but is still incompletely understood is the association between HSV infection and AD. On a population level, data from a Swedish cohort suggest that reactivation of HSV-1, indicated by high serum levels of anti-HSV immunoglobulin M (IgM) antibodies, increases the risk of developing AD[55]. In vitro experiments have shown that HSV-1 induces increases in intracellular amyloid-β (Aβ) levels and tau phosphorylation, both of which are markers of AD[56]. In addition, HSV-1 DNA has been found in amyloid plaques in patients with AD, further underlining a link between the virus and pathological aggregations[57]. Virus-mediated cytotoxicity does not explain all clinical developments in AD, but viruses and amyloid plaques are thought to alter the neuroimmune crosstalk in various ways. For example, HSV-1 proteins debilitate neuronal autophagy and, consequently, antigen presentation[58]. In mice, recurrent asymptomatic activation of HSV-1 leads to upregulation of markers of neuroinflammation (for example, Toll-like recep-tor-4, IFNα and IFNβ) and early neurodegeneration[59]. In addition, extracellular deposits of Aβ activate resting microglia and trigger them to attack neurons via various pathways, such as NADPH oxidase activation, inducible nitric oxide synthase expression and phagocytosis[60].
In summary, owing to the sensitive equilibrium of the immune system and brain function, infectious agents might tip the scales towards pathology. Furthermore, infections can trigger autoimmune phenomena that might contribute to a broad spectrum of neuropsychiatric diseases, discussed in more detail below.
04. Autoimmunity¶
04.00.¶
Nonspecific inflammatory processes underlie the low-grade inflammation described above, but neuro-inflammation can also be driven by autoimmunity. Autoimmune responses can be antibody driven or cell driven and, depending on the autoantigen involved, can involve antigen presentation by major histocompatibility complex (MHC) class II. Autoimmune responses can follow infection, and molecular mimicry can underlie these responses[41],61. Innate and adaptive immune mecha nisms have roles in autoimmunity. What we know about autoimmune responses and neuro psychiatric disorders is largely based on specific disorders, discussed in detail below.
04.01. Lessons from multiple sclerosis¶
Neurological autoimmune diseases are mainly considered to be mediated by autoaggressive lymphocytes or anti-neuronal or anti-glial autoantibodies. These conditions include neuro myelitis optica and potentially disabling, or sometimes life-threatening, autoimmune encephalomyelitis. MS is the classical and most common chronic autoimmune encephalomyelitis; this disease leads to demyelination and progressive neurodegeneration. In early MS, most patients experience transient physical deficits, such as optic neuritis, hemiparesis or sensory disturbances, in a relapsing–remitting presentation. Although these physical deficits are the most obvious consequences, an increasing body of evidence illustrates that cognitive deficits are abundant and extensive in MS[62].
Patients with MS can experience neuropsychological symptoms, such as mild cognitive alterations or depression, related not only to relapses but also to phases of remission[63]. The most common cognitive deficits, which occur in 40–60% of patients with MS, are reduced processing speed and impaired memory and/or executive function, and these symptoms can improve to some extent with immunomodulatory treatment[64]. Behavioural signs, such as anxiety, can also occur and are currently seen as comorbidities but might be a consequence of the pathology[65]. Notably, cognitive impairment at the time of MS diagnosis predicts disability progression, transition to secondary progressive MS and cortical thinning66. In line with these findings, the risk of cognitive impairment increases with progression of disease and is high-est in secondary progressive MS[67]. Nevertheless, 30–40% of patients with clinically isolated syndrome, which is considered to be a precursor of MS, have cognitive deficits[68],[69], and in a study of so-called benign MS (defined by an Expanded Disability Status Scale (EDSS) score ≤3.0 despite a disease duration ≥15 years), up to 45% of patients had cognitive impairment[70].
Inflammation in MS, the local effects of which include microglial activation and myelin and neuronal damage, is thought to be primarily mediated by auto reactive T cells. Pro-inflammatory TH17 cells are thought to migrate into the CNS and subsequently increase BBB permeability, enabling invasion of other immune cells[71] (FIG. 1b). Other important players include regulatory T cells, which are functionally impaired in people with MS[72], and B cells, the role of which is highlighted by the effects of B cell-depleting therapies[73]. Relapses in patients with MS are influenced by stressful life events and by infections: a systematic meta-analysis has shown that upper respiratory tract infections have the most pronounced effect on relapse rates74. These observations underline the role of inflammation in relapse activity, as both systemic infection and stress induce a pro-inflammatory status, as discussed in more detail below.
A specific autoantigen has not yet been identified in MS, but autoreactive T cells are widely assumed to target proteins of the myelin sheath, causing demyelination and, thereby, white matter damage. Demyelination deprives neurons of protective factors and has been described to decrease axonal transport and synaptic density in demyelinated hippocampi from post-mortem MS brains[75]. However, direct recognition of neurons by patho genic T cells has also been proposed[76],[77], and neuronal loss could occur as a result of Wallerian degene ration and mitochondrial dysfunction[78]. Grey matter damage, measured as the cortical lesion load or cortical thinning, is a key predictor of progressive disease and cognitive decline[79]. Even in clinically stable patients, the presence of gadolinium-enhancing lesions on brain MRI is associated with impaired performance in the Paced Auditory Serial Addition Test (PASAT), a screening tool for cognitive dysfunction[80]. These findings underline the fact that active neuroinflammation detectable with gadoli nium enhancement affects cognitive function and might cause detrimental effects on neuronal networks and connectivity.
Although rare, psychotic symptoms can also occur in MS. A genetic overlap between MS and schizophrenia has been identified, and the genes that are common to both conditions are immune related81. Despite being separate disease entities, MS and classical psychiatric disorders are accompanied by higher-order network disturbances82,83,[84],[85] (FIG. 2a). The study of MS has provided fundamental insights into the crosstalk between the immune system and the nervous system, and these insights will be highly valuable for the study of other diseases in which underlying immune pathology has recently been identified, discussed below.
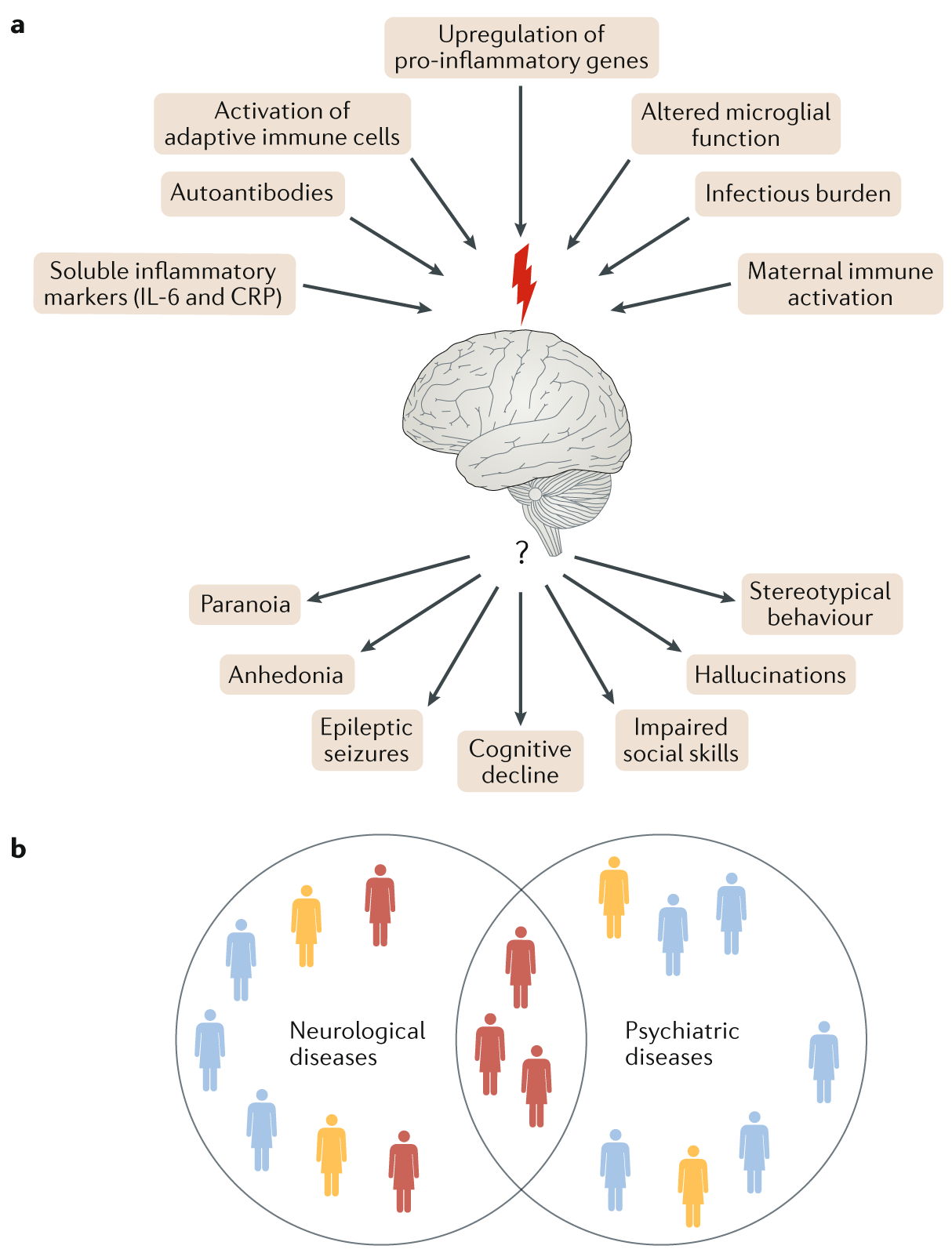
Fig. 2 | Overlap of neuropsychiatric disorders.¶
a | Higher-order network disturbances are caused by common pathophysiological mechanisms of immune dysregulation (top). How clinically distinct phenotypes emerge from these pathways is an unresolved question (bottom).
b | As pathways emerge that are common to neurological and psychiatric autoimmune brain diseases, the traditional boundaries between these disorders become blurred. Identification of subgroups of patients who are likely to benefit from immunotherapeutic approaches is crucial. CRP, C-reactive protein.
04.02. Lessons from autoimmune psychosis¶
HSV encephalitis
- autoAb against:
- GABAAR
- AMPA (subunit GluA2)
A compelling example of the overlap between psychiatric and neuro-logical pathologies is the occurrence of psychosis as a result of autoantibodies against neuropil, which can develop spontaneously in neoplastic diseases or after viral infections, particularly HSV encephalitis[86]. Some of these autoantibodies target intracellular antigens, such as onconeural proteins or 65 kDa glutamic acid decarboxylase (GAD65). The presence of these antibodies is diagnostically useful but considered to be an epiphenomenon, as cytotoxic T cells are responsible for neuronal damage that is associated with neuropsy-chiatric symptoms87. However, a broad spectrum of extracellular antigens involved in synaptic transmission and plasticity are located on the cell surface; therefore, autoantibodies against these antigens that are present in patients with psychosis seem highly likely to have a direct pathogenic role. One example of such an anti-body is the autoantibody against the GABAA receptor, a postsynaptic chloride channel that mediates fast inhibitory neurotransmission in the mammalian brain; this autoantibody causes encephalitis with therapy-resistant epileptic seizures[88]. Moreover, antibodies against the α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA) receptor can have a pathological role: administration of human pathogenic antibodies against the AMPA receptor subunit GluA2 impairs long-term synaptic plasticity in vitro and affects learning and memory in mice in vivo[89].
NMDA
More controversial is the importance of antibodies against the N-methyl-D-aspartate (NMDA) glutamate receptor, one of the most commonly observed antigens in autoimmune encephalitis. Patients with this condition often present with anxiety, sleep disorders, mania, paranoia, memory impairment and disintegration of language, followed by a phase in which agitation and catatonia alternate, accompanied by abnormal movements and autonomic instability[90]. In humans, antibody titres in the CSF correlate with the clinical course of disease[90]. Experiments in rats indicate a pathophysiological role for anti-NMDA receptor antibodies. For example, in rat hippocampal neurons that have been incubated with anti-NMDA receptor antibodies, NMDA receptor internalization and reduced NMDA receptor-mediated synaptic currents were observed[91]. In addition, treatment of rats with NMDA receptor blockers evoked stereotyped behaviour and cataleptic freezing that were comparable to symptoms of autoimmune encephalitis in humans[92]. NMDA receptor encephalitis can occur after HSV encephalitis in up to 27% of patients, indicating a role for molecular mimicry61. Furthermore, antibodies that cross-react with the NMDA receptor have been found in patients with neuropsychiatric systemic lupus erythematosus, and these antibodies cause neuronal damage via activated microglia and complement component C1q when administered to mice[93],[94].
Despite this evidence, the pathological role of neurotransmitter receptor autoantibodies came into question when anti-NMDA receptor autoantibodies were detected in serum from ~10% of healthy controls as well as in patients with pure psychotic symptoms[95]. Similar results have been seen for a variety of brain antigens, challenging the pathological relevance of CNS-specific autoantibodies[96]; BBB integrity has been proposed as a pivotal factor in the clinical outcome[95]. In another study, anti-NMDA receptor autoantibodies from psychotic patients altered synaptic transmission and long-term potentiation in cultured neurons and in mouse brain whereas those from healthy controls did not97. Independent of immune pathogenesis, glutamate receptor hypofunction has been proposed as a key factor in the development of schizophrenia[98].
In combination, these results provide evidence that specific and different mechanisms underlie encephalitis and autoimmune psychosis, in which autoantibodies against NMDA receptors have different effects on the organization of the glutamate synapse through receptor internalization or abnormal NMDA receptor dynamics. Given that patients with schizophrenia who are positive for anti-NMDA receptor antibodies do not exhibit classical signs of encephalitis97, we conclude that different immune alterations mediated by similar antibodies result in different neuropsychiatric entities.
05. Immunopsychiatry - an emerging field¶
On the basis of the data discussed above, the concept of autoimmune psychosis has become a compelling example of the interface between neurological (autoimmune encephalitis) and psychiatric (psychosis) disorders. In this section, we discuss accumulating evidence that suggests that immune dysregulation is involved in a broad spectrum of psychiatric diseases. Owing to a lack of specific markers, studies in the emerging field of immunopsychiatry currently focus on systemic measures of inflammation. A growing number of studies have identified nonspecific inflammatory disturbances in subgroups of patients, but knowledge of specific pathogenic pathways remains scarce.
One overarching concept is that immunopsychiatric diseases involve a generally overactivated immune system. In general, overactivation of the immune system is thought to increase brain vulnerability, increasing the risk of psychiatric symptoms upon a so-called second hit later in life99. On an epidemiological level, this hypothesis is supported by a link between psychiatric diseases, such as schizophrenia, depression and anxiety, and systemic autoimmune disorders[100],[101]. Case reports similarly support this hypothesis; one striking example is the development of severe psychosis in an individual who received a stem cell transplantation from their brother with schizophrenia, indicating that adoptive immune transfer had a role[102]. Furthermore, extensive studies of cytokine profiles in disorders such as schizophrenia, depression, suicidal ideation and post-traumatic stress disorder reveal wide-ranging dysregulation of mainly pro-inflammatory markers, in particular IL-6, IL-2 receptor, IL-1β, IL-17 A and C-reactive protein (CRP)[103]–[107]. Several studies identified a positive correlation of serum levels of inflammatory markers with disease severity and a negative correlation with cognitive performance[103],[108].
Changes in the innate CNS immune compartment have also been associated with psychiatric disorders. Either excessive upregulation or downregulation of microglia function evokes detrimental effects. For example, genetic defects in microglial signalling pathways cause abnormal development of brain circuits and neuro-psychiatric symptoms, as in hereditary diffuse leukoencephalopathy with spheroids (HDLS)[109]. Disruption of microglial function in later life can be initiated by psycho social and environmental factors. For example, in a mouse model, chronic stress with depressive behaviour has been associated with microglial loss in the hippocampus[110]. By contrast, depression and schizophrenia have been associated with increased microglial activity measured with PET, and this increased activity could be used to identify patients at high risk of disease exacerbations[111],[112]. Although microglial activation in humans has been studied only indirectly, early life challenges are assumed to ‘prime’ microglia and increase their response to subsequent inflammatory stimuli[113].
A crucial discovery that many immune factors are influential prenatally led to use of the so-called maternal immune activation model in order to study ASD and schizophrenia in rodents. In this model, IL-6 was identified as a key mediator of inflammatory effects on fetal brain development[114]. In mice, autism-like behaviour in offspring requires the presence of mater-nal RORγt-positive TH17 cells and IL-17a downstream of IL-6 (REF.[115]). In subsequent studies, a mechanism of ASD pathogenesis has been proposed in which TH17-dependent loss of inhibitory interneuron networks leads to increased cortical activation in the primary somatosensory cortex[116].
A study published in 2017 provides evidence that maternal gut commensal bacteria have a role in IL-17a production in mothers[117]. These findings support the idea of a gut–immune–brain axis that has been implicated in classical autoimmunity (such as modulation of the balance between pro-inflammatory and regulatory T cells by gut bacteria in MS[118]) and are in line with previous reports of an altered intestinal barrier in people with ASD and their close relatives[119]. In rodents, the role of maternal immune activation in ASD was confirmed at the transcriptome level, in which immune activation in the mother caused dysregulation in the fetal brain expression of genes involved in developmental processes[120]. Although animal studies provide the proof of concept and agree with findings in diseases of neurological immune dysfunction, such as MS, the mechanisms described are plausible in humans but remain speculative.
Cytokine signalling is not the only aspect of maternal immunity that affects the developing fetal brain — B cell-mediated immune responses and production of antibodies can also have effects. For example, in an epidemio logical study, antibodies against fetal brain tissue were detected in the serum of mothers of children with ASD but not in mothers of healthy children[121]. Another study produced the same observation in up to 23% of mothers of children with ASD[122]. In animal studies, intravenous administration of 73 kDa and 37 kDa IgG from mothers of children with ASD to pregnant rhesus macaques led to abnormal social behaviour in the macaque offspring[123]. Targets for these antibodies include proteins with functions in neurodevelopment, such as contactin-associated protein-like 2 (CASPR2)[122],[124]. Although the exact mecha nism by which these maternal antibodies affect the fetus is not yet understood, the findings in humans and the adoptive transfer study suggest that placental transfer of maternal antibodies contributes to disease development, at least in a subgroup of patients.
Aberrant genetic regulation underlies most of the immune dysfunction discussed above, and a polygenic contribution to the risk of several severe psychiatric disorders has been confirmed in a meta-analysis of genome-wide association studies in which the MHC was the most relevant shared risk locus[125]. Genes in the MHC are involved in physiological processes, including CNS development and homeostasis, but are common among neuropsychiatric disease-associated loci[126],[127]. For example, the MHC-resident complement C4 cluster has been implicated in the risk of schizophrenia[128],[129]. Therefore, fine dissection of the genetic contribution of the MHC is a promising approach to increasing our understanding of brain disorders.
Although we are only beginning to develop a deeper, mechanistic understanding of immunological pathways involved in the pathogenesis of psychiatric disorders, the emerging field of immunopsychiatry provides new perspectives on the disorders discussed. As even early evolutionary processes such as the integration of sequences of human endogenous retroviruses into the genome presumably through repeated infections were found to be associated with neurological and psychiatric disorders[130],[131], it becomes clear that dysfunctional processes occur on an ancestral, maternal and individual level.
06. Immunomodulatory treatment¶
Classical neurological autoimmune diseases have long been treated with immunomodulatory drugs. Routinely used treatments for acute exacerbations include steroids, plasmapheresis, intravenous immunoglobulin, cyclo-phosphamide and B cell-depleting monoclonal anti-bodies; a broadening range of disease-modifying drugs is emerging from the development of therapies for MS (BOX 2). Immunosuppression is also an established treat-ment strategy for autoimmune encephalitis, but clinical studies of such treatments for psychiatric diseases are still in progress.
In a pilot study published in 2018, treatment with high-dose intravenous immunoglobulin had positive effects on scores in several cognitive and behavioural tests for children with ASD and evidence of a dysregu-lated immune system132. In another study, intravenous immunoglobulin had beneficial effects for subgroups of children with ASD and inflammation133. A series of case reports of immune modulatory treatment in PANDAS disorders demonstrates how an understanding that auto-inflammatory processes are involved in the patho-genesis can lead to successful treatment134. However, syste matic treatment approaches are still lacking and are currently merely performed with antibiotics.
Currently, monoclonal antibody treatments are being trialled in schizophrenia. In one study, use of the mono clonal antibody natalizumab, which targets the cell adhesion molecule α4 integrin and is currently used for treatment of MS, is being tested135. The recombinant humanized anti-human IL-6 receptor monoclonal anti-body tocilizumab is being tested in two136,137 ongoing studies, one of which136 focuses on patients with signs of peripheral inflammation. In one case report, remis-sion of treatment-resistant schizophrenia was seen in a patient who underwent bone marrow transplantation for acute myeloid leukaemia, providing evidence that this treatment could be an effective, or even curative, option for severe schizophrenia138. Similarly, infusion of autologous umbilical cord blood into children with ASD led to clinical improvement of core autistic symptoms139.
Evidence that immune dysfunction is involved in depression is supported by accumulating data that sug-gest that established antidepressant drugs such as selec-tive serotonin reuptake inhibitors (SSRIs) exert their effects, at least partly, via anti-inflammatory effects. For example, a meta-analysis showed that SSRIs reduce levels of IL-1β and IL-6 in patients with depression140. In line with these observations, synthetic metabolites of tryptophan, a precursor of serotonin, suppressed pro-inflammatory T cells and autoimmune neuro-inflammation in experimental autoimmune encephalitis, a mouse model of MS141. Additional support for the con-cept that immune pathology underlies depression comes from a meta-analysis of randomized clinical trials that showed that, despite heterogeneous study results, non-steroidal anti-inflammatory drugs had an overall positive effect on depression142. In rats, the microglia inhibitor minocycline improved symptoms of depression in the forced swimming test143, an observation that is consistent with reports that microglia are overactivated in depres-sion112. A large randomized trial of minocycline has not been done in humans, but a meta-analysis of existing clinical data indicates beneficial effects of mino cycline in patients with depression, thereby providing a proof of concept144. Minocycline also had positive effects as an adjunctive treatment to risperidone in children with ASD145, although not as a single medication in a small pilot study146. In addition to its inhibitory effects on microglia, minocycline has antibiotic properties that might modify the gut flora, which could contri bute to beneficial treatment outcomes. Furthermore, in depres-sion, anti-inflammatory treatment with the TNF antago-nist infliximab had beneficial effects in patients with treatment-resistant depression and high baseline levels of inflammatory biomarkers147.
In summary, immunomodulatory therapies are effec-tive options for a range of neurological and psychiatric diseases. To develop individualized therapeutic strate-gies, subgroups of patients with psychiatric disorders and signs of immune dysregulation must be identified (FIG. 2b). Clinical studies are more likely to reveal con-sistent positive effects if they include patients with an inflammatory phenotype, indicated by MRI or high levels of inflammation markers in the CSF or serum. Furthermore, we assume that there is a narrow thera-peutic time window for maximal therapeutic efficacy and avoidance of irreversible CNS injury. For example, timely immunomodulatory treatment in autoimmune encephalitis probably prevents neuronal damage and permanent neurological deficits148, and this concept might hold true for other neuropsychiatric disorders.
Box 02. Immunomodulatory drugs available for the treatment of CNS autoimmune disorders¶
Immunomodulatory drugs and treatments applied in a broad range of autoimmune diseases
- Steroids
- Plasmapheresis
- Intravenous immunoglobulin
- Cyclophosphamide
- Methotrexate
- Mycophenolate mofetil
- Azathioprin
- Mitoxantrone
- Rituximab
- Ocrelizumab
- Infliximab
Disease-modifying drugs from the range of therapeutics for multiple sclerosis
- IFNβ
- Glatiramer acetate
- Teriflunomide
- Dimethyl fumarate
- Fingolimod
- Natalizumab
- Alemtuzumab
- Cladribine
- Rituximab
- Ocrelizumab
Other drugs with partial immunomodulatory effects
- Nonsteroidal anti-inflammatory drugs
- Selective serotonin reuptake inhibitors
- Minocycline
- Tryptophan metabolites
07. Resilience and reserve¶
A well-recognized risk factor for exacerbation in MS and many psychiatric disorders is psychosocial stress. Some evidence suggests that the association between psychiatric disorders and stress is mediated, at least in part, by neuroinflammatory processes, including microglial activation. For example, the ‘two-hit hypo-thesis’ proposes that stress in early life increases base-line microglial activity, thereby increasing the risk of psychiatric disorders upon subsequent challenges later in life149,150. Accordingly, a meta-analysis of clinical stud-ies demonstrated a pro-inflammatory status (high levels of CRP, IL-6 and TNF) in adults with a history of child-hood trauma151. Similarly, in a prospective cohort study, patients with a history of childhood maltreatment and depression had higher levels of CRP than participants with depression only152.
The question remains, however, of why some indi-viduals are more resilient to stressful events than others. In a mouse model of chronic social defeat stress, BBB integrity was altered in the nucleus accumbens owing to downregulation of the tight junction protein claudin 5, thereby allowing influx of IL-6 into the brain paren-chyma153. Importantly, however, loss of claudin 5 was observed only in stress-susceptible mice — susceptibil-ity to stress was observed as social avoidance behaviour after several days of exposure to social defeat by a larger, physically aggressive mouse; by contrast, this behaviour was not seen in mice classified as resilient. Epigenetic modulation of IL-6 seems to further promote resilience in mice, as an inhibitor of IL-6 DNA methylation that decreased IL-6 production reduced depressive symptoms in a mouse model154. Furthermore, adaptive immune cells are needed for coping with stress. For example, exposure of mice to acute psychological stress in the form of predator odour increases T cell trafficking to the brain155, and T cell-deficient mice displayed more signs of maladaptation after stress156. In addition, analogous to findings for neurogenesis discussed above, self-reactive, brain-specific T cells are needed for resilience in mice, leading to the idea that vaccination with CNS-related peptides could stimulate coping behaviour157.
From studies of chronic autoimmune inflammation of the CNS in MS, we know that the brain has reserve capacities30. Our recent studies indicate that the cortical network responds to an inflammatory attack with a TNF-dependent increase in cortical synaptic strength65. This upregulation of cortical neuronal activity presum-ably reflects repair mechanisms initially but eventually causes maladaptive development of anxiety behaviour despite otherwise complete remission in disability. Analysis of higher-order networks in patients who progress rapidly and those who do not could increase our understanding of brain reserve and identify future therapeutic approaches.
08. New clinical approach¶
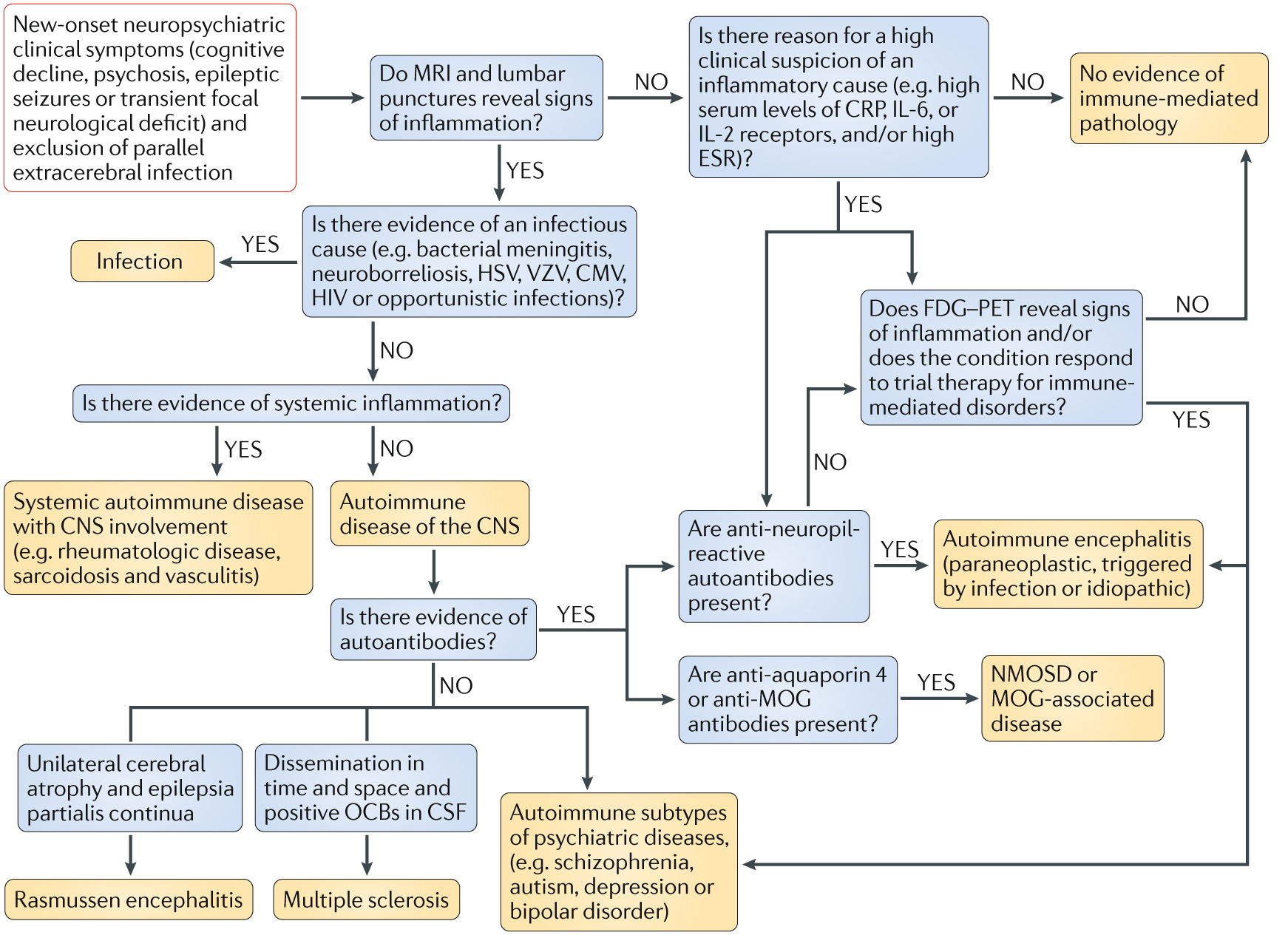
Fig. 3 | A proposed clinical pathway for patients with new-onset neuropsychiatric symptoms.¶
Blue boxes indicate investigations required and orange boxes indicate diagnoses. CMV, cytomegalovirus; CRP, C-reactive protein; CSF, cerebrospinal fluid; ESR , erythrocyte sedimentation rate; HSV, herpes simplex virus; MOG, myelin oligodendrocyte glycoprotein; NMOSD, neuromyelitis optica spectrum disorder ; OCB, oligoclonal bands; VZV, varicella zoster virus.
¶
The idea that the CNS is a site of immune privilege has shifted with a growing understanding that immune cells have multifaceted roles in CNS homeostasis, even in the healthy brain. Mechanistic aspects of the neuroim-mune interplay have been revealed, including roles for peripherally induced adaptive immunity and for cross-talk between neurons and CNS-resident microglia. The involvement of astrocytes has been studied to a lesser extent but is becoming a focus of research.
Associations of the immune system with brain dis-eases have been reported for decades, initially in case reports but later in epidemiological studies. Studies of the immune system in psychiatric disorders have thus far focused on classical markers of inflammation, such as CRP and erythrocyte sedimentation rate (ESR), TNF or IL-6 in serum, as more sensitive or specific markers are often lacking. A level of inflammation that strikes a highly sensitive equilibrium seems to be required for health. Consequently, any disturbance can cause detrimental effects not only on focal neurological symptoms but also, more importantly, on higher-order network function.
As common immunological aetiologies of neurologi-cal and psychiatric diseases are discovered, the traditional classification of these diseases is increasingly challenged. In order to develop promising immune-based thera-peutic approaches for psychiatric diseases, we propose new diagnostic pathways (FIG. 3). In this proposed algo-rithm, individuals who present with new neuropsy-chiatric symptoms, such as cognitive and behavioural disturbances, epileptic seizures and psychosis, must be screened for blood, CSF and imaging markers of inflammation. Psychiatric symptoms in patients with classical neuroinflammatory disorders must be con-sidered when making treatment decisions. If there are any signs of active inflammation, immunomodulatory treatment should be discussed.
09. Conclusion¶
For decades, or even centuries, of medical history, neuro-psychiatric diseases have been diagnosed and classified on the basis of phenomenological criteria. The findings discussed in this Review provide novel perspectives and indicate the need for a future classification of neuropsy-chiatric disorders on the basis of pathogenic mechanisms, at least for subgroups of patients. In this context, diag-nostic algorithms should classify patients into inflamma-tory and non-inflammatory subgroups to help reach an individualized treatment decision. For example, future understanding of neuropsychiatric diseases might no longer divide them into descriptive entities, such as MS, neuromyelitis optica spectrum disorder, auto immune encephalitis, schizophrenia, ASD and bipolar dis order, but into pathogenic entities, such as autoimmune antibody-mediated or T cell-mediated brain disorders.
Although the knowledge of immune involvement in neuropsychiatric disorders has emerged over several years, necessary changes to the clinical management of patients with these disorders have scarcely been imple-mented. Furthermore, many questions remain unsolved (BOX 3), such as why apparently similar mechanisms lead to clinically distinct disorders and whether immunolog-ical disturbances are always a cause of certain disorders or sometimes a consequence. The next steps will require combined experience from neurologists and psychiatrists in a new field of immunoneuropsychiatry.
Box 03. Unresoloved questions to direct future reseach¶
- Which pathways mediate the interplay between levels of inflammation and cognitive performance?
- Why do apparently similar immunological mechanisms lead to clinically distinct disorders?
- What are the precise mechanisms of interaction between immune cells and higher-order networks in neuropsychiatric diseases? How can this crosstalk be addressed therapeutically?
- Are immunological disturbances the cause or consequence of neuropsychiatric diseases?
- How can we better identify subgroups of patients who are likely to benefit from immunotherapies?
10. References¶
- [Nimmerjahn, A., Kirchhoff, F. & Helmchen, F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 308, 1314–1318 (2005).][1]
- [Stevens, B. et al. The classical complement cascade mediates CNS synapse elimination. Cell 131, 1164–1178 (2007).][2]
- [Paolicelli, R. C. et al. Synaptic pruning by microglia is necessary for normal brain development. Science 333, 1456–1458 (2011).][3]
- Sanchez-Alcaniz, J. A. et al. Cxcr7 controls neuronal migration by regulating chemokine responsiveness. Neuron 69, 77–90 (2011).
- [Wake, H., Moorhouse, A. J., Jinno, S., Kohsaka, S. & Nabekura, J. Resting microglia directly monitor the functional state of synapses in vivo and determine the fate of ischemic terminals. J. Neurosci. 29, 3974–3980 (2009).][5]
- [Pocock, J. M. & Kettenmann, H. Neurotransmitter receptors on microglia. Trends Neurosci. 30, 527–535 (2007).][6]
- [Antonucci, F. et al. Microvesicles released from microglia stimulate synaptic activity via enhanced sphingolipid metabolism. EMBO J. 31, 1231–1240 (2012).][7]
- [Beattie, E. C. et al. Control of synaptic strength by glial TNFα. Science 295, 2282–2285 (2002).][8]
- Gertig, U. & Hanisch, U. K. Microglial diversity by responses and responders. Front. Cell Neurosci. 8, 101 (2014).
- Ellwardt, E., Walsh, J. T., Kipnis, J. & Zipp, F. Understanding the role of T cells in CNS homeostasis. Trends Immunol. 37, 154–165 (2016).
- [Engelhardt, B. & Ransohoff, R. M. Capture, crawl, cross: the T cell code to breach the blood-brain barriers. Trends Immunol. 33, 579–589 (2012).][11]
- [Schwartz, M. & Deczkowska, A. Neurological disease as a failure of brain-immune crosstalk: the multiple faces of neuroinflammation. Trends Immunol. 37, 668–679 (2016).][12]
- Strominger, I. et al. The choroid plexus functions as a niche for T-cell stimulation within the central nervous system. Front. Immunol. 9, 1066 (2018).
- [Filiano, A. J. et al. Unexpected role of interferon- gamma in regulating neuronal connectivity and social behaviour. Nature 535, 425–429 (2016).][14]
- [Derecki, N. C. et al. Regulation of learning and memory by meningeal immunity: a key role for IL-4. J. Exp. Med. 207, 1067–1080 (2010).][15]
- Kipnis, J., Cohen, H., Cardon, M., Ziv, Y. & Schwartz, M. T cell deficiency leads to cognitive dysfunction: implications for therapeutic vaccination for schizophrenia and other psychiatric conditions. Proc. Natl Acad. Sci. USA 101, 8180–8185 (2004).
This study shows that a lack of mature T cells causes cognitive and behavioural impairment in mice, underlining the role of adaptive immunity for normal CNS function. - [Ziv, Y. et al. Immune cells contribute to the maintenance of neurogenesis and spatial learning abilities in adulthood. Nat. Neurosci. 9, 268–275 (2006).][17]
- [Walsh, J. T. et al. MHCII-independent CD4+ T cells protect injured CNS neurons via IL-4. J. Clin. Invest. 125, 699–714 (2015).][18]
- Vogelaar, C. F. et al. Fast direct neuronal signaling via the IL-4 receptor as therapeutic target in neuroinflammation. Sci. Transl Med. 10, eaao2304 (2018).
This study reveals a beneficial effect of intrathecal IL-4 treatment on progression in experimental autoimmune encephalomyelitis, thereby highlighting a new pathway of neuroimmune interplay as a potential therapeutic target. - [Radjavi, A., Smirnov, I. & Kipnis, J. Brain antigen-reactive CD4+ T cells are sufficient to support learning behavior in mice with limited T cell repertoire. Brain Behav. Immun. 35, 58–63 (2014).][20]
- [Iliff, J. J. et al. A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid beta. Sci. Transl Med. 4, 147ra111 (2012).][21]
- Xie, L. et al. Sleep drives metabolite clearance from the adult brain. Science 342, 373–377 (2013).
- Louveau, A. et al. Structural and functional features of central nervous system lymphatic vessels. Nature 523, 337–341 (2015).
This article presents the first description of the re-discovery of the meningeal lymphatic system. - [Absinta, M. et al. Human and nonhuman primate meninges harbor lymphatic vessels that can be visualized noninvasively by MRI. eLife 6, e29738 (2017).][24]
- [Traka, M., Podojil, J. R., McCarthy, D. P., Miller, S. D. & Popko, B. Oligodendrocyte death results in immune-mediated CNS demyelination. Nat. Neurosci. 19, 65–74 (2016).][25]
- D’Mello, C. & Swain, M. G. Immune-to-brain communication pathways in inflammation-associated sickness and depression. Curr. Top. Behav. Neurosci. 31, 73–94 (2017).
- Kelley, K. W. et al. Cytokine-induced sickness behavior. Brain Behav. Immun. 17 (Suppl. 1), 112–118 (2003).
- [Neilley, L. K., Goodin, D. S., Goodkin, D. E. & Hauser, S. L. Side effect profile of interferon beta-1b in MS: results of an open label trial. Neurology 46, 552–554 (1996).][28]
- [Tarr, A. J., Liu, X., Reed, N. S. & Quan, N. Kinetic characteristics of euflammation: the induction of controlled inflammation without overt sickness behavior. Brain Behav. Immun. 42, 96–108 (2014).]][29]
- Larochelle, C., Uphaus, T., Prat, A. & Zipp, F. Secondary progression in multiple sclerosis: neuronal exhaustion or distinct pathology? Trends Neurosci. 39, 325–339 (2016).
- Franceschi, C. et al. Inflamm-aging. An evolutionary perspective on immunosenescence. Ann. NY Acad. Sci. 908, 244–254 (2000).
- Marrie, R. A. et al. Increased incidence of psychiatric disorders in immune-mediated inflammatory disease. J. Psychosom. Res. 101, 17–23 (2017).
- Perry, V. H., Nicoll, J. A. & Holmes, C. Microglia in neurodegenerative disease. Nat. Rev. Neurol. 6, 193–201 (2010).
- Lumeng, C. N. & Saltiel, A. R. Inflammatory links between obesity and metabolic disease. J. Clin. Invest. 121, 2111–2117 (2011).
- Stranahan, A. M., Hao, S., Dey, A., Yu, X. & Baban, B. Blood-brain barrier breakdown promotes macrophage infiltration and cognitive impairment in leptin receptor-deficient mice. J. Cereb. Blood Flow Metab. 36, 2108–2121 (2016).
- Guillemot-Legris, O. & Muccioli, G. G. Obesity-induced neuroinflammation: beyond the hypothalamus. Trends Neurosci. 40, 237–253 (2017).
- Benros, M. E. et al. Autoimmune diseases and severe infections as risk factors for schizophrenia: a 30-year population-based register study. Am. J. Psychiatry 168, 1303–1310 (2011). This large Danish longitudinal register shows that both autoimmune disease and prior hospitalization for infection increase the risk of schizophrenia on an epidemiological level.
- Khandaker, G. M. et al. Inflammation and immunity in schizophrenia: implications for pathophysiology and treatment. Lancet Psychiatry 2, 258–270 (2015).
- Canetta, S. et al. Elevated maternal C-reactive protein and increased risk of schizophrenia in a national birth cohort. Am. J. Psychiatry 171, 960–968 (2014).
- Atladottir, H. O. et al. Maternal infection requiring hospitalization during pregnancy and autism spectrum disorders. J. Autism Dev. Disord. 40, 1423–1430 (2010).
- Orlovska, S. et al. Association of streptococcal throat infection with mental disorders: testing key aspects of the PANDAS hypothesis in a nationwide study. JAMA Psychiatry 74, 740–746 (2017).
- Zomorrodi, A. & Wald, E. R. Sydenham’s chorea in western Pennsylvania. Pediatrics 11 7, e675–e679 (2006).
- Swedo, S. E. et al. Pediatric autoimmune neuropsychiatric disorders associated with streptococcal infections: clinical description of the first 50 cases. Am. J. Psychiatry 155, 264–271 (1998).
- Bronze, M. S. & Dale, J. B. Epitopes of streptococcal M proteins that evoke antibodies that cross-react with human brain. J. Immunol. 151, 2820–2828 (1993).
- Singer, H. S., Gause, C., Morris, C. & Lopez, P. Serial immune markers do not correlate with clinical exacerbations in pediatric autoimmune neuropsychiatric disorders associated with streptococcal infections. Pediatrics 121, 1198–1205 (2008).
- US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT02889016 (2016).
- Kapur, N. et al. Herpes simplex encephalitis: long term magnetic resonance imaging and neuropsychological profile. J. Neurol. Neurosurg. Psychiatry 57, 1334–1342 (1994).
- Almanzar, G. et al. Long-term cytomegalovirus infection leads to significant changes in the composition of the CD8+ T cell repertoire, which may be the basis for an imbalance in the cytokine production profile in elderly persons. J. Virol. 79, 3675–3683 (2005).
- Katan, M. et al. Infectious burden and cognitive function: the Northern Manhattan Study. Neurology 80, 1209–1215 (2013). This cohort study shows that infectious burden measured as serological exposure to common pathogens was associated with cognitive impairment independent of cardiovascular risk profile.
- Hamdani, N. et al. Effects of cumulative herpesviridae and Toxoplasma gondii infections on cognitive function in healthy, bipolar, and schizophrenia subjects. J. Clin. Psychiatry 78, e18–e27 (2017).
- Kamminga, J., Cysique, L. A., Lu, G., Batchelor, J. & Brew, B. J. Validity of cognitive screens for HIV-associated neurocognitive disorder: a systematic review and an informed screen selection guide. Curr. HIV/AIDS Rep. 10, 342–355 (2013).
- Zhang, C. J. et al. TLR-stimulated IRAKM activates caspase-8 inflammasome in microglia and promotes neuroinflammation. J. Clin. Invest. 128, 5399–5412 (2018).
- Heneka, M. T., McManus, R. M. & Latz, E. Inflammasome signalling in brain function and neurodegenerative disease. Nat. Rev. Neurosci. 19, 610–621 (2018).
- Wendeln, A. C. et al. Innate immune memory in the brain shapes neurological disease hallmarks. Nature 556, 332–338 (2018). This study shows that peripheral inflammatory stimuli induce differential epigenetic modulation of brain-resident microglia, influencing symptoms in a mouse model of AD; these findings provide a mechanistic link between inflammation, innate immunity and neuropsychiatric disease.
- Lovheim, H., Gilthorpe, J., Adolfsson, R., Nilsson, L. G. & Elgh, F. Reactivated herpes simplex infection increases the risk of Alzheimer’s disease. Alzheimers Dement. 11 , 593–599 (2015).
- Fulop, T., Itzhaki, R. F., Balin, B. J., Miklossy, J. & Barron, A. E. Role of microbes in the development of Alzheimer’s disease: state of the art - an International Symposium presented at the 2017 IAGG Congress in San Francisco. Front. Genet. 9, 362 (2018).
- Wozniak, M. A., Mee, A. P. & Itzhaki, R. F. Herpes simplex virus type 1 DNA is located within Alzheimer’s disease amyloid plaques. J. Pathol. 217, 131–138 (2009).
- O’Connell, D. & Liang, C. Autophagy interaction with herpes simplex virus type-1 infection. Autophagy 12, 451–459 (2016).
- Martin, C. et al. Inflammatory and neurodegeneration markers during asymptomatic HSV-1 reactivation. J. Alzheimers Dis. 39, 849–859 (2014).
- Brown, G. C. & Neher, J. J. Inflammatory neurodegeneration and mechanisms of microglial killing of neurons. Mol. Neurobiol. 41, 242–247 (2010).
- Armangue, T. et al. Frequency, symptoms, risk factors, and outcomes of autoimmune encephalitis after herpes simplex encephalitis: a prospective observational study and retrospective analysis. Lancet Neurol. 17, 760–772 (2018).
This combined prospective observational study and retrospective analysis characterizes the frequency and the natural course of autoimmune encephalitis after herpes simplex encephalitis. - Carta, M. G. et al. The risk of bipolar disorders in multiple sclerosis. J. Affect. Disord. 155, 255–260 (2014).
- Feinstein, A., Magalhaes, S., Richard, J. F., Audet, B. & Moore, C. The link between multiple sclerosis and depression. Nat. Rev. Neurol. 10, 507–517 (2014).
- Patti, F. Treatment of cognitive impairment in patients with multiple sclerosis. Expert Opin. Investig. Drugs 21, 1679–1699 (2012).
- [Ellwardt, E. et al. Maladaptive cortical hyperactivity upon recovery from experimental autoimmune encephalomyelitis. Nat. Neurosci. 21, 1392–1403 (2018).
This study identifies the emergence of cortical network hyperexcitability and elevated anxiety in remission after neuroinflammatory attack to the brain; this network instability with anxiety behaviour, also known in patients with MS, represents the early stages of neurodegeneration.][65] - Pitteri, M., Romualdi, C., Magliozzi, R., Monaco, S. & Calabrese, M. Cognitive impairment predicts disability progression and cortical thinning in MS: an 8-year study. Mult. Scler. 23, 848–854 (2017).
- Potagas, C. et al. Cognitive impairment in different MS subtypes and clinically isolated syndromes. J. Neurol. Sci. 267, 100–106 (2008).
- Ruano, L. et al. Age and disability drive cognitive impairment in multiple sclerosis across disease subtypes. Mult. Scler. 23, 1258–1267 (2017).
- Di Filippo, M., Portaccio, E., Mancini, A. & Calabresi, P. Multiple sclerosis and cognition: synaptic failure and network dysfunction. Nat. Rev. Neurosci. 19, 599–609 (2018).
- Amato, M. P. et al. Benign multiple sclerosis: cognitive, psychological and social aspects in a clinical cohort. J. Neurol. 253, 1054–1059 (2006).
- Nylander, A. & Hafler, D. A. Multiple sclerosis. J. Clin. Invest. 122, 1180–1188 (2012).
- Viglietta, V., Baecher-Allan, C., Weiner, H. L. & Hafler, D. A. Loss of functional suppression by CD4+CD25+ regulatory T cells in patients with multiple sclerosis. J. Exp. Med. 199, 971–979 (2004).
- Hauser, S. L. et al. Ocrelizumab versus interferon beta-1a in relapsing multiple sclerosis. N. Engl. J. Med. 376, 221–234 (2017).
- McKay, K. A., Jahanfar, S., Duggan, T., Tkachuk, S. & Tremlett, H. Factors associated with onset, relapses or progression in multiple sclerosis: a systematic review. Neurotoxicology 61, 189–212 (2017).
- Dutta, R. et al. Demyelination causes synaptic alterations in hippocampi from multiple sclerosis patients. Ann. Neurol. 69, 445–454 (2011).
- Liblau, R. S., Gonzalez-Dunia, D., Wiendl, H. & Zipp, F. Neurons as targets for T cells in the nervous system. Trends Neurosci. 36, 315–324 (2013).
- Kebir, H. et al. Human TH17 lymphocytes promote blood-brain barrier disruption and central nervous system inflammation. Nat. Med. 13, 1173–1175 (2007).
- Campbell, G. R. et al. Mitochondrial DNA deletions and neurodegeneration in multiple sclerosis. Ann. Neurol. 69, 481–492 (2011).
- Calabrese, M. et al. Cortical lesion load associates with progression of disability in multiple sclerosis. Brain 135, 2952–2961 (2012).
- Bellmann-Strobl, J. et al. Poor PASAT performance correlates with MRI contrast enhancement in multiple sclerosis. Neurology 73, 1624–1627 (2009).
- Andreassen, O. A. et al. Genetic pleiotropy between multiple sclerosis and schizophrenia but not bipolar disorder: differential involvement of immune-related gene loci. Mol. Psychiatry 20, 207–214 (2015).
- Charalambous, T. et al. Structural network disruption markers explain disability in multiple sclerosis. J. Neurol. Neurosurg. Psychiatry 90, 219–226 (2019).
- Fleischer, V. et al. Graph theoretical framework of brain networks in multiple sclerosis: a review of concepts. Neuroscience https://doi.org/10.1016/ j.neuroscience.2017.10.033 (2017).
- Dong, D., Wang, Y., Chang, X., Luo, C. & Yao, D. Dysfunction of large-scale brain networks in schizophrenia: a meta-analysis of resting-state functional connectivity. Schizophr. Bull. 44, 168–181 (2018).
- Kaiser, R. H., Andrews-Hanna, J. R., Wager, T. D. & Pizzagalli, D. A. Large-scale network dysfunction in major depressive disorder: a meta-analysis of resting-state functional connectivity. JAMA Psychiatry 72, 603–611 (2015).
- Armangue, T. et al. Autoimmune post-herpes simplex encephalitis of adults and teenagers. Neurology 85, 1736–1743 (2015).
- Bien, C. G. et al. Immunopathology of autoantibody-associated encephalitides: clues for pathogenesis. Brain 135, 1622–1638 (2012).
- Petit-Pedrol, M. et al. Encephalitis with refractory seizures, status epilepticus, and antibodies to the GABAA receptor: a case series, characterisation of the antigen, and analysis of the effects of antibodies. Lancet Neurol. 13, 276–286 (2014).
- Haselmann, H. et al. Human autoantibodies against the AMPA receptor subunit GluA2 induce receptor reorganization and memory dysfunction. Neuron 100, 91–105 (2018).
- Dalmau, J., Lancaster, E., Martinez-Hernandez, E., Rosenfeld, M. R. & Balice-Gordon, R. Clinical experience and laboratory investigations in patients with anti-NMDAR encephalitis. Lancet Neurol. 10, 63–74 (2011).
- Hughes, E. G. et al. Cellular and synaptic mechanisms of anti-NMDA receptor encephalitis. J. Neurosci. 30, 5866–5875 (2010).
- Haggerty, G. C., Forney, R. B. & Johnson, J. M. The effect of a single administration of phencyclidine on behavior in the rat over a 21-day period. Toxicol. Appl. Pharmacol. 75, 444–453 (1984).
- DeGiorgio, L. A. et al. A subset of lupus anti-DNA antibodies cross-reacts with the NR2 glutamate receptor in systemic lupus erythematosus. Nat. Med. 7, 1189–1193 (2001).
- Nestor, J. et al. Lupus antibodies induce behavioral changes mediated by microglia and blocked by ACE inhibitors. J. Exp. Med. 215, 2554–2566 (2018).
- Hammer, C. et al. Neuropsychiatric disease relevance of circulating anti-NMDA receptor autoantibodies depends on blood-brain barrier integrity. Mol. Psychiatry 19, 1143–1149 (2014).
- Dahm, L. et al. Seroprevalence of autoantibodies against brain antigens in health and disease. Ann. Neurol. 76, 82–94 (2014).
- Jezequel, J. et al. Dynamic disorganization of synaptic NMDA receptors triggered by autoantibodies from psychotic patients. Nat. Commun. 8, 1791 (2017). This study reveals differential effects of autoantibodies against the glutamate NMDA receptor on synaptic transmission and long-term potentiation in patients with psychosis versus healthy controls, providing a mechanistic framework for different clinical outcomes despite similar autoantibodies
- Kantrowitz, J. & Javitt, D. C. Glutamatergic transmission in schizophrenia: from basic research to clinical practice. Curr. Opin. Psychiatry 25, 96–102 (2012).
- Bergink, V., Gibney, S. M. & Drexhage, H. A. Autoimmunity, inflammation, and psychosis: a search for peripheral markers. Biol. Psychiatry 75, 324–331 (2014).
- Cullen, A. E. et al. Associations between non- neurological autoimmune disorders and psychosis: a meta-analysis. Biol. Psychiatry 85, 35–48 (2018).
- Siegmann, E. M. et al. Association of depression and anxiety disorders with autoimmune thyroiditis: a systematic review and meta-analysis. JAMA Psychiatry 75, 577–584 (2018).
- Sommer, I. E. et al. Severe chronic psychosis after allogeneic SCT from a schizophrenic sibling. Bone Marrow Transplant. 50, 153–154 (2015).
- Dahan, S. et al. The relationship between serum cytokine levels and degree of psychosis in patients with schizophrenia. Psychiatry Res. 268, 467–472 (2018).
- Haapakoski, R., Mathieu, J., Ebmeier, K. P., Alenius, H. & Kivimaki, M. Cumulative meta-analysis of interleukins 6 and 1β, tumour necrosis factor α and C-reactive protein in patients with major depressive disorder. Brain Behav. Immun. 49, 206–215 (2015).
- Black, C. & Miller, B. J. Meta-analysis of cytokines and chemokines in suicidality: distinguishing suicidal versus nonsuicidal patients. Biol. Psychiatry 78, 28–37 (2015).
- Al-Ayadhi, L. Y. & Mostafa, G. A. Elevated serum levels of interleukin-17A in children with autism. J. Neuroinflamm. 9, 158 (2012).
- Passos, I. C. et al. Inflammatory markers in post-traumatic stress disorder: a systematic review, meta-analysis, and meta-regression. Lancet Psychiatry 2, 1002–1012 (2015).
- Bulzacka, E. et al. Chronic peripheral inflammation is associated with cognitive impairment in schizophrenia: results from the multicentric FACE-SZ dataset. Schizophr. Bull. 42, 1290–1302 (2016).
- Rademakers, R. et al. Mutations in the colony stimulating factor 1 receptor (CSF1R) gene cause hereditary diffuse leukoencephalopathy with spheroids. Nat. Genet. 44, 200–205 (2011).
- Tong, L. et al. Microglia loss contributes to the development of major depression induced by different types of chronic stresses. Neurochem. Res. 42, 2698–2711 (2017).
- Bloomfield, P. S. et al. Microglial activity in people at ultra high risk of psychosis and in schizophrenia: an [11C]PBR28 PET brain imaging study. Am. J. Psychiatry 173, 44–52 (2016). This PET imaging study shows that patients with schizophrenia and individuals with a high risk of psychosis exhibit increased microglial activity, indicating a connection between neuroinflammation and the risk of psychosis.
- Wachholz, S. et al. Microglia activation is associated with IFN-alpha induced depressive-like behavior. Brain Behav. Immun. 55, 105–113 (2016).
- Norden, D. M., Muccigrosso, M. M. & Godbout, J. P. Microglial priming and enhanced reactivity to secondary insult in aging, and traumatic CNS injury, and neurodegenerative disease. Neuropharmacology 96, 29–41 (2015).
- Smith, S. E., Li, J., Garbett, K., Mirnics, K. & Patterson, P. H. Maternal immune activation alters fetal brain development through interleukin-6. J. Neurosci. 27, 10695–10702 (2007).
- Choi, G. B. et al. The maternal interleukin-17a pathway in mice promotes autism-like phenotypes in offspring. Science 351, 933–939 (2016). The results of this study indicate that the development of ASD-like phenotypes in offspring in the murine model of maternal immune activation is mediated by TH17 cells.
- Shin Yim, Y. et al. Reversing behavioural abnormalities in mice exposed to maternal inflammation. Nature 549, 482–487 (2017).
- Kim, S. et al. Maternal gut bacteria promote neurodevelopmental abnormalities in mouse offspring. Nature 549, 528–532 (2017). This study shows that the production of IL-17 in the maternal immune activation model in mice depends on the composition of maternal intestinal bacteria, underlining the role of the gut–immune–brain axis.
- Cekanaviciute, E. et al. Gut bacteria from multiple sclerosis patients modulate human T cells and exacerbate symptoms in mouse models. Proc. Natl Acad. Sci. USA 114 , 10713–10718 (2017).
- de Magistris, L. et al. Alterations of the intestinal barrier in patients with autism spectrum disorders and in their first-degree relatives. J. Pediatr. Gastroenterol. Nutr. 51, 418–424 (2010).
- Lombardo, M. V. et al. Maternal immune activation dysregulation of the fetal brain transcriptome and relevance to the pathophysiology of autism spectrum disorder. Mol. Psychiatry 23, 1001–1013 (2018).
- Zimmerman, A. W. et al. Maternal antibrain antibodies in autism. Brain Behav. Immun. 21, 351–357 (2007).
- Braunschweig, D. et al. Autism-specific maternal autoantibodies recognize critical proteins in developing brain. Transl Psychiatry 3, e277 (2013).
- Bauman, M. D. et al. Maternal antibodies from mothers of children with autism alter brain growth and social behavior development in the rhesus monkey. Transl Psychiatry 3, e278 (2013).
- Brimberg, L. et al. Caspr2-reactive antibody cloned from a mother of an ASD child mediates an ASD-like phenotype in mice. Mol. Psychiatry 21, 1663–1671 (2016). This study describes the isolation and characterization of monoclonal brain-reactive antibodies from a mother of a child with ASD.
- Cross-Disorder Group of the Psychiatric Genomics Consortium. Identification of risk loci with shared effects on five major psychiatric disorders: a genome-wide analysis. Lancet 381, 1371–1379 (2013).
- Trowsdale, J. & Knight, J. C. Major histocompatibility complex genomics and human disease. Annu. Rev. Genomics Hum. Genet. 14, 301–323 (2013).
- Dendrou, C. A., Petersen, J., Rossjohn, J. & Fugger, L. HLA variation and disease. Nat. Rev. Immunol. 18, 325–339 (2018).
- Sekar, A. et al. Schizophrenia risk from complex variation of complement component 4. Nature 530, 177–183 (2016).
- Prasad, K. M. et al. Neuropil contraction in relation to Complement C4 gene copy numbers in independent cohorts of adolescent-onset and young adult-onset schizophrenia patients-a pilot study. Transl Psychiatry 8, 134 (2018).
- Kury, P. et al. Human endogenous retroviruses in neurological diseases. Trends Mol. Med. 24, 379–394 (2018).
- Perron, H. et al. Molecular characteristics of human endogenous retrovirus type-W in schizophrenia and bipolar disorder. Transl Psychiatry 2, e201 (2012).
- Melamed, I. R., Heffron, M., Testori, A. & Lipe, K. A pilot study of high-dose intravenous immunoglobulin 5% for autism: impact on autism spectrum and markers of neuroinflammation. Autism Res. 11, 421–433 (2018).
- Connery, K. et al. Intravenous immunoglobulin for the treatment of autoimmune encephalopathy in children with autism. Transl Psychiatry 8, 148 (2018). This article and that by Melamed et al. (2018) describe pilot studies that provided the first evidence for a beneficial effect of intravenous immunoglobulin for the treatment of children with ASD and signs of inflammation.
- Swedo, S. E., Frankovich, J. & Murphy, T. K. Overview of treatment of pediatric acute-onset neuropsychiatric syndrome. J. Child Adolesc. Psychopharmacol. 27, 562–565 (2017).
- US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT03093064 (2017).
- US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT02874573 (2018).
- US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT02034474 (2018).
- Miyaoka, T. et al. Remission of psychosis in treatment- resistant schizophrenia following bone marrow transplantation: a case report. Front. Psychiatry 8, 174 (2017).
- Dawson, G. et al. Autologous cord blood infusions are safe and feasible in young children with autism spectrum disorder: results of a single-center phase I open-label trial. Stem Cells Transl Med. 6, 1332–1339 (2017).
- Hannestad, J., DellaGioia, N. & Bloch, M. The effect of antidepressant medication treatment on serum levels of inflammatory cytokines: a meta-analysis. Neuropsychopharmacology 36, 2452–2459 (2011).
- Platten, M. et al. Treatment of autoimmune neuroinflammation with a synthetic tryptophan metabolite. Science 310, 850–855 (2005).
- Kohler, O. et al. Effect of anti-inflammatory treatment on depression, depressive symptoms, and adverse effects: a systematic review and meta-analysis of randomized clinical trials. JAMA Psychiatry 71, 1381–1391 (2014).
- Molina-Hernandez, M., Tellez-Alcantara, N. P., Perez-Garcia, J., Olivera-Lopez, J. I. & Jaramillo-Jaimes, M. T. Antidepressant-like actions of minocycline combined with several glutamate antagonists. Prog. Neuropsychopharmacol. Biol. Psychiatry 32, 380–386 (2008).
- Rosenblat, J. D. & McIntyre, R. S. Efficacy and tolerability of minocycline for depression: a systematic review and meta-analysis of clinical trials. J. Affect. Disord. 227, 219–225 (2018). This systematic review and meta-analysis provides a proof of concept for the antidepressant effects of minocycline.
- Ghaleiha, A. et al. Minocycline as adjunctive treatment to risperidone in children with autistic disorder: a randomized, double-blind placebo-controlled trial. J. Child Adolesc. Psychopharmacol. 26, 784–791 (2016).
- Pardo, C. A. et al. A pilot open-label trial of minocycline in patients with autism and regressive features. J. Neurodev. Disord. 5, 9 (2013).
- Raison, C. L. et al. A randomized controlled trial of the tumor necrosis factor antagonist infliximab for treatment-resistant depression: the role of baseline inflammatory biomarkers. JAMA Psychiatry 70, 31–41 (2013).
- Kalman, B. Autoimmune encephalitides: a broadening field of treatable conditions. Neurologist 22, 1–13 (2017).
- Howes, O. D. & McCutcheon, R. Inflammation and the neural diathesis-stress hypothesis of schizophrenia: a reconceptualization. Transl Psychiatry 7, e1024 (2017).
- Fonken, L. K., Frank, M. G., Gaudet, A. D. & Maier, S. F. Stress and aging act through common mechanisms to elicit neuroinflammatory priming. Brain Behav. Immun. 73, 133–148 (2018).
- Baumeister, D., Akhtar, R., Ciufolini, S., Pariante, C. M. & Mondelli, V. Childhood trauma and adulthood inflammation: a meta-analysis of peripheral C-reactive protein, interleukin-6 and tumour necrosis factor-α. Mol. Psychiatry 21, 642–649 (2016).
- Danese, A. et al. Elevated inflammation levels in depressed adults with a history of childhood maltreatment. Arch. Gen. Psychiatry 65, 409–415 (2008).
- Menard, C. et al. Social stress induces neurovascular pathology promoting depression. Nat. Neurosci. 20, 1752–1760 (2017). This study reveals mechanistic links between psychosocial stress and depression via disruption of BBB integrity by downregulation of claudin 5 in mice.
- Wang, J. et al. Epigenetic modulation of inflammation and synaptic plasticity promotes resilience against stress in mice. Nat. Commun. 9, 477 (2018).
- Lewitus, G. M., Cohen, H. & Schwartz, M. Reducing post-traumatic anxiety by immunization. Brain Behav. Immun. 22, 1108–1114 (2008).
- Cohen, H. et al. Maladaptation to mental stress mitigated by the adaptive immune system via depletion of naturally occurring regulatory CD4+CD25+ cells. J. Neurobiol. 66, 552–563 (2006).
- Lewitus, G. M. et al. Vaccination as a novel approach for treating depressive behavior. Biol. Psychiatry 65, 283–288 (2009).
¶